Stresshormonregulation und Depressionsrisiko – Perspektiven für die antidepressive Behandlung
Forschungsbericht (importiert) 2011 - Max Planck Institut für Psychiatrie
Depression – eine Stresserkrankung
Vielfältige Befunde weisen darauf hin, dass Stress und dessen neurobiologische Korrelate bei Entwicklung, Auslösung und Verlauf der Depression eine wesentliche Rolle spielen. So zeigen die Ergebnisse epidemiologischer Längsschnittstudien, dass frühe soziale Belastungsfaktoren, etwa Vernachlässigung, körperliche Gewalt oder sexueller Missbrauch, aber auch die Auswirkungen von Naturkatastrophen oder anderen Umwelt-Traumata das Risiko deutlich erhöhen, als Erwachsener an einer Depression zu erkranken. Auch im Vorfeld des Ausbruchs einer depressiven Episode treten gehäuft belastende Ereignisse auf. Insbesondere Veränderungen des persönlichen Alltags gelten als Risikofaktoren für depressive Episoden. Diese umfassen Änderungen der Lebensumstände aufgrund von Ausbildung und Beruf, Veränderungen in der Partnerschaft oder der Familie, Schwangerschaft, Verlust von Bezugspersonen, Überlastung oder Ausgrenzungen.
Das wichtigste neuroendokrine Korrelat der Stressreaktion beim Menschen ist das „Stresshormon“ Cortisol, dessen Ausschüttung durch die Hypothalamus-Hypophysen-Nebennierenrinden-Achse (englisch: hypothalamus-pituitary-adrenocortical, HPA) gesteuert wird. Unterschiedliche Arten von Stress führen gleichermaßen zur Aktivierung von Neuronen des paraventrikulären Nukleus des Hypothalamus und damit zur Synthese der Neuropeptide Corticotropin Releasing Hormone (CRH) und Vasopressin (AVP, für Arginin-Vasopressin, der humanen Variante dieses Neuropeptids), die über den Portalblutkreislauf zum Vorderlappen der Hypophyse gelangen. Dort stimulieren sie die Synthese und Ausschüttung des adrenocorticotropen Hormons (ACTH), das wiederum Biosynthese und Freisetzung von Cortisol aus der Nebennierenrinde aktiviert.
 – Achse erfolgt in erster Linie über Glucocorticoidrezeptoren. Bei vielen depressiven Patienten besteht eine Insensitivität dieser Rezeptoren, die zu einer Verstellung des Regulationsgleichgewichts im HPA-System führt.")
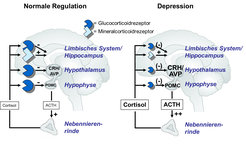
Cortisol hat eine Vielzahl von Funktionen, die für die Anpassung des Organismus an eine akute Stresssituation (über)lebensnotwendig sind. So führt es über den Mechanismus der Gluconeogenese zu einer Mobilisierung von Energieressourcen; gleichzeitig werden kurzfristig entbehrliche Funktionen der zellulären und humoralen Immunabwehr herunterreguliert. Damit stehen dem Organismus erhöhte Energiereserven zur Verfügung, um die Stresssituation zu bewältigen. Nach Beendigung der Stresssituation oder nach Anpassung des Individuums an die geänderten Bedingungen ist eine schnelle Rückregulation der HPA-Achse erforderlich. Geschieht dies nicht, beispielsweise aufgrund chronischer Belastung, kann es zu vielerlei Störungen kommen, etwa im Stoffwechsel, der Immunabwehr, im Herz/Kreislaufsystem, zu Lern- und Gedächtnisstörungen und schließlich auch zur Depression. Folglich ist eine fein abgestimmte Anpassungsfähigkeit der Stresshormonregulation wesentlich, um die Entwicklung einer pathologischen Regulationsstörung zu vermeiden. Die wichtigsten regulatorischen Elemente sind die Glucocorticoid- und Mineralcorticoidrezeptoren. Insbesondere der Glucocorticoidrezeptor (GR) ist für die Rückregulation der HPA-Achse verantwortlich (Abb. 1). Dies geschieht über negative Rückkopplung, sodass aufgrund der Stresseinwirkung erhöhte Cortisolspiegel gleichzeitig zur Dämpfung der Neuropeptide CRH und AVP (auf hypothalamischer Ebene) und des Peptidhormons ACTH (auf hypophysärer Ebene) beitragen.
, haben im weiteren Behandlungsverlauf die deutlich günstigere Prognose (helle Verlaufslinie, Kreise) im Vergleich zu Patienten, bei denen keine Verbesserung der HPA-Achsenregulation erzielt werden konnte (eingebettetes Bild rechts oben; dunkle Verlaufslinie, Dreiecke) (modifiziert nach [1]).")

Die Regulationsfähigkeit der HPA-Achse kann mithilfe des sog. Dexamethason (Dex)/CRH-Tests überprüft werden. Dabei wird das Wechselspiel zwischen dem dämpfenden Dex-Effekt und der stimulierenden CRH-Wirkung überprüft. Bei optimaler HPA-Achsenregulation kommt es zu einer Supprimierung der Cortisolsekretion, die trotz CRH-Gabe nur unwesentlich ansteigt. Bei einer Fehlregulation der HPA-Achse kommt es zu einer verminderten Cortisolsuppression, gefolgt von einer deutlichen Cortisolstimulation durch CRH. Ursachen für diese Fehlregulation sind eine abgeschwächte Sensitivität der Glucocorticoidrezeptoren sowie eine Überaktivität der hypothalamischen Neuropeptide CRH und AVP. Bei vielen depressiven Patienten findet sich in der akuten Episode eine gestörte Stresshormonregulation (Abb. 1). Bei rezidivierenden Formen der Depression wird dies besonders häufig festgestellt. Unter antidepressiver Behandlung kommt es jedoch in den meisten Fällen zu einer Normalisierung der Regulationsstörung. Im Dex/CRH-Test drückt sich dies in einer verminderten Cortisolantwort aus, wenn der Test nach erfolgreicher Behandlung wiederholt wird. Die Normalisierung der HPA-Achsenregulation scheint dabei dem Therapieerfolg vorauszugehen. So zeigt ein Teil der Patienten eine rasche Verbesserung der gestörten HPA-Achsenregulation unter antidepressiver Behandlung, während bei anderen Patienten trotz intensiver Therapie keine Änderung der gestörten HPA-Achsenregulation festzustellen ist. Letztere haben eine ungünstige Prognose in Hinblick auf den weiteren Behandlungsverlauf unter der aktuellen Therapie [1]. Die Verbesserung der gestörten HPA-Achsenregulation scheint daher eine notwendige Voraussetzung für den antidepressiven Therapieerfolg zu sein (Abb. 2).
Genetik der Stresshormonregulation
 Variationen im FKBP5-Gen (hier: rs1360780) beeinflussen die Protein-konzentration von FKBP51, dem Expressionsprodukt dieses Gens. (B) Personen mit dem TT-Genotyp des dargestellten FKBP5-Polymorphismus zeigen eine verstärkte Cortisolreaktion unter psychosozialem Stress und insbesondere eine verlangsamte und unvollständige Rückregulation des Stresshormons nach Ende der Belastung, auch bei einer Wiederholung der Stresssituation. Dies bestätigt die funktionell GR-antagonistischen Eigenschaften von FKBP51 und weist auf dessen klinische Relevanz in Hinblick auf eine beeinträchtigte Stresshormonregulation hin (modifiziert nach [2], [3]).")

Neben aktuellen und biografischen Umweltfaktoren des Individuums beeinflussen auch genetische Faktoren die Stresshormonregulation. Aufgrund seiner Bedeutung für die HPA-Achsenregulation stehen der Glucocorticoidrezeptor und seine funktionsvermittelnden Hilfsmoleküle, die sog. Chaperone, im Mittelpunkt der genetischen Stressforschung. Um seine regulierende Funktion zu entfalten, muss der aktivierte Rezeptor in den Zellkern eindringen, wo er an spezifizierten Bindungsstellen der zellulären DNA bindet und die Transkription nachfolgend regulierter Gene beeinflusst. Neben genetischen Variationen im NR3C1-Gen, das den GR exprimiert, sind insbesondere Varianten im FKBP5-Gen für die Stresshormonregulation von Bedeutung. FKBP5 exprimiert das Protein FKBP51 (FK506-Binding-Protein 5), ein sog. Immunophilin, dessen Bezeichnung von seiner Bindungseigenschaft für FK506, einen immunsuppressiven Arzneistoff, herrührt. FKBP51 gehört zu den GR-Chaperonmolekülen. Es wirkt als Hemmer der GR-Bindungsfähigkeit für Cortisol (und andere Glucocorticoide) und behindert die GR-Translokation, d.h. die Wanderung des aktivierten GR in den Zellkern. FKBP51 ist daher ein funktioneller Antagonist des Glucocorticoidrezeptors.
Variationen im FKBP5-Gen beeinflussen die Expression von FKBP51 [2]. Die Träger des seltenen Allels des FKBP5-Polymorphismus rs1360780 weisen erhöhte FKBP51-Konzentrationen im Blut auf (Abb. 3A). Aufgrund der funktionell GR-antagonistischen Eigenschaften von FKBP51 beeinträchtigt dies die Regulationsfähigkeit der HPA-Achse. Das hat zur Folge, dass Träger dieser genetischen Variante eine verstärkte Cortisolreaktion unter psychosozialem Stress aufweisen [3]. Insbesondere die Rückregulation der HPA-Achse ist verlangsamt und bleibt für längere Zeit unvollständig, auch bei wiederholter Stressexposition (Abb. 3B).
FKBP5-Genomik und Depression
Aufgrund der funktionalen Bedeutung des FKBP5-Gens für die Stresshormonregulation liegt es nahe, einen Bezug zwischen genetischen FKBP5-Variationen und dem Erkrankungsrisiko für Depression zu vermuten. Depression ist jedoch eine multifaktorielle Erkrankung. Genetische Faktoren tragen etwa 40 Prozent zum Erkrankungsrisiko bei. Weitere krankheitsdeterminierende Faktoren sind in der individuellen Biografie der Patienten zu suchen. Insbesondere Stressfaktoren, wie belastende Umweltereignisse, spielen hierbei eine wichtige Rolle, wie eingangs bereits dargestellt. Aufgrund dieses Umstands ist nicht zu erwarten, dass eine einzelne genetische Variation das Depressionsrisiko entscheidend beeinflusst. Viel wahrscheinlicher ist, dass Wechselwirkungen zwischen der genetischen Disposition und Belastungen aufgrund von Umweltfaktoren das Depressionsrisiko determinieren. Dies konnte jüngst für Variationen im FKBP5-Gen in einer prospektiv-epidemiologischen Studie am Max-Planck-Institut für Psychiatrie nachgewiesen werden [4]. Träger des FKBP5-Genotyps, für den eine erhöhte FKBP51-Aktivität nachgewiesen wurde (rs1360780 TT, Abb. 3A), weisen dann ein deutlich erhöhtes Depressionsrisiko auf, wenn sie traumatisierenden Ereignissen, etwa körperlicher Gewalt, sexuellem Missbrauch oder schweren Unfällen, ausgesetzt waren. Sind solche belastenden Ereignisse nicht eingetreten, weisen die Probanden trotz Risikogenotyps keine erhöhte Depressionswahrscheinlichkeit auf. Vergleichbare Ergebnisse konnten für weitere Varianten des FKBP5-Gens gezeigt werden, etwa für rs3800373 (Abb. 4).
.")
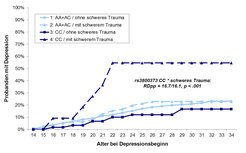
Diese Ergebnisse basieren auf einer prospektiv-epidemiologischen Datenanalyse. Dadurch ist gewährleistet, dass die identifizierten traumatischen Ereignisse tatsächlich vor Ausbruch der Depression aufgetreten sind und nicht etwa durch eine retrospektive Erinnerungsverzerrung, die bei Depression vielfach beschrieben ist, beeinflusst sind. Dies ist eine wichtige Voraussetzung, um eine kausale Verknüpfung zwischen der FKBP51-vermittelten genetischen Regulationsschwäche der HPA-Achse und einem erhöhten Depressionsrisiko nach traumatischen Belastungen postulieren zu können.
Die beschriebenen genetischen Variationen im FKBP5-Gen beeinflussen nicht nur das Depressionsrisiko, sondern auch den antidepressiven Behandlungserfolg bei Patienten [2]. Dies führte zu der Annahme, dass FKBP51 als Expressionsprodukt von FKBP5 eine wichtige Rolle auch bei der Depressionsbehandlung spielt. Vorläufige Befunde, die am Max-Planck-Institut für Psychiatrie bei Patienten mit Depression erhoben wurden, bestätigen dies: Patienten, bei denen sich unter antidepressiver Behandlung eine Reduktion der FKBP51-Expression einstellt, zeigen im Dex/CRH-Test eine stärkere Reduktion der Cortisolantwort und haben einen günstigeren Therapieverlauf als Patienten, bei denen keine Reduzierung der FKBP51-Expresson festgestellt wurde. Die Reduktion der FKBP51-Expression scheint somit die Normalisierung einer gestörten HPA-Achsenregulation zu begünstigen und dadurch den Therapieverlauf zu verbessern.
Perspektiven für die antidepressive Behandlung
In den letzten Jahren und Jahrzehnten gab es zwar vielerlei Fortschritte in Bezug auf Verträglichkeit und Nebenwirkungsspektrum von Antidepressiva, ihre therapeutische Effizienz verbesserte sich jedoch kaum. Betrachtet man die pharmakologischen Wirkmechanismen, kann dies nicht überraschen. Nahezu alle antidepressiven Arzneimittel bewirken direkt oder indirekt eine Verstärkung der monoaminergen Neurotransmission. Dies steht im Kontrast zur Vielschichtigkeit des depressiven Symptombilds, das sich in einer großen Variationsbreite der individuellen Depression ausdrückt. Für eine verbesserte Effektivität der Depressionsbehandlung scheint es daher unumgänglich, die individuelle Depressionspathologie zu berücksichtigen. Dieser Ansatz der sog. Personalisierten Medizin erfordert die Entwicklung neuer Antidepressiva mit spezifischem Wirkmechanismus sowie von Biomarkern, die die Patienten mit der passenden Pathologie identifizieren können [5]. Die Ergebnisse zur Stresshormonregulation können hierzu einen wichtigen Beitrag leisten. Mit dem Dex/CRH-Test steht ein sensitiver Biomarker zur Identifizierung von Patienten mit einer Regulationsstörung der HPA-Achse zur Verfügung. Für diese Patienten könnte eine spezifische Intervention mit dem Ziel der Hemmung der FKBP51-Aktivität zu einem deutlich schnelleren antidepressiven Therapieerfolg führen als dies mit den gegenwärtig verfügbaren monoaminergen Antidepressiva alleine der Fall ist. Die Arbeitsgruppe Chemische Genomik am Max-Planck-Institut für Psychiatrie arbeitet derzeit intensiv an der Entwicklung selektiver Hemmstoffe von FKBP51. Erste vielversprechende Ergebnisse zum Entwicklungskonzept wurden jüngst publiziert [6].