
Research Group Molecular Neurogenetics
Jan Deussing
Genetic risk factors
, gain-of-function (green) or neutral (gray) polymorphisms as revealed by various in vitro assays.")
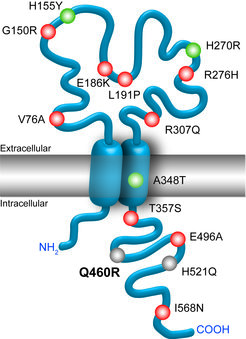
The causal underpinnings of psychiatric disorders are largely unknown. The identification of susceptibility genes has been challenging with many discoveries failing replication even in the era of genome-wide association studies (GWAS). This disappointment is largely owing to the inherent phenotypic and genetic heterogeneity of these disorders as well as to the difficulty to control for environmental factors interfering with disease etiology. However, increasing sample sizes as well as prospective whole genome sequencing efforts will increase the statistical power. These approaches have the potential to identify more reliable candidate risk factors, affected pathways and even rare coding variants of bigger effect size.
Despite the above mentioned difficulties, numerous genetic variants associated with psychiatric disorders have been identified by human genetic studies in recent years (Fig. 1). A current limitation is that most of the discovered disease-associated genetic variants lack experimental validation in vivo in a suitable animal model. Therefore, we aim to address the contribution of candidate genes to the predisposition to disease development, in particular in conjunction with stress as an environmental factor. The establishment of appropriate genetic mouse models will aid the understanding of the impact of these putative susceptibility genes on the pathoetiology of psychiatric disorders. Moreover, these mouse models will help to elucidate the potential of these candidate genes as biomarkers or novel pharmacological targets for therapeutic intervention.
Epigenetic mechanisms
 are subjected to numerous post-translational modifications at their N-terminal tail which protrudes from the nucleosome. These histone modifications directly regulate the chromatin structure and thus gene expression. For simplification only those lysine residues are highlighted, which are subjected to acetylation or deactylation by histone acetyltransferases (HATs) and histone deacetylases (HDACs), respectively. Many of these lysine residues can also prone to methylation (Me).")
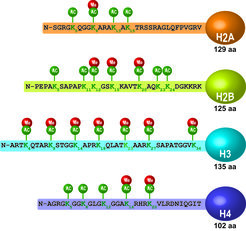
Despite compelling epidemiological evidence for the heritability of psychiatric disorders, only a small number of genetic risk factors has been identified to date. This phenomenon termed as “missing heritability” might to some extent be explained by limitations of current genetic approaches (see above). However, growing evidence suggests that epigenetic mechanisms might play an important role through which environmental exposures directly interact with the individual´s genetic composition to determine the disease risk. Epigenetics refers to stable changes in gene transcription that are based on alterations in the chromatin structure (e.g. DNA methylation or histone modifications) independent of any modification of the primary DNA sequence.
There is ample evidence that altered chromatin plasticity is involved in psychiatric diseases. For instance, histone deacetylase (HDAC) inhibitors can improve depression-related symptoms in humans and in corresponding mouse models. Moreover, stressors have been shown to directly affect the epigenome by changing histone acetylation (Fig. 2). These observations open new avenues for a better understanding of the pathophysiology of stress-related disorders. Therefore, we aim to determine effects of HDACs on emotional behavior, cognition and the response to stress. These behavioral readouts are correlated with changes in the transcriptome and genome-wide acetylation signatures in the brain to better understand potential protective or detrimental effects of HDACs in the stress response.
Neural circuits of stress
Besides genetic and epigenetic factors, disturbances of neural circuits – the functional units of brain cells that mediate complex processes such as thoughts, feelings or behavior - are likely to be involved in neuropsychiatric disorders. Moreover, neural circuits are closely regulated by genetic and epigenetic factors, which might directly impact on circuit development, structure, functional integrity and dynamics. The corticotropin-releasing hormone (CRH) system represents the prototype of a stress circuit and is perfectly situated as a mediator of gene × environment interactions. The maladaptation of the CRH system has been causally linked to stress-related neuropsychiatric pathologies such as mood and anxiety disorders. CRH plays a critical role in integrating the neuroendocrine, behavioral and autonomic responses to stress. To coordinate these purposes, CRH possesses a dual function acting as a secretagogue within the context of the hypothalamic-pituitary-adrenal (HPA) axis and as a neuroregulator at extrahypothalamic sites.
 family of neuropeptides comprises four family members: CRH, Urocortin 1 (UCN1), UCN2 and UCN3. The ligands have different affinities towards the CRH receptors (CRHR1, CRHR2 and soluble variants of CRHR2) and the CRH binding protein (CRHBP). CRH-related neuropeptides and their cognate receptors are expressed in specific patterns throughout the brain. Depicted is the distribution on a sagittal section of a mouse brain.")
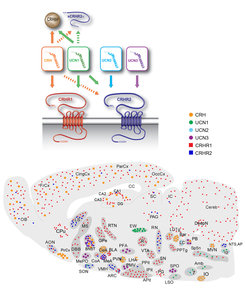
While the CRH-dependent control of the neuroendocrine stress response is well understood, we have only just begun to understand the complexity of extrahypothalamic CRH/CRH receptor (CRHR) circuits that control behavioral stress responses (Fig. 3). We use mouse genetic tools to address the in vivo function of the components of the CRH system, i.e. the ligands (CRH, UCN1, UCN2, UCN3) and their receptors (CRHR1, CRHR2). Applying conditional approaches (spatially or temporally controlled), we can selectively interrogate neural circuit-specific functions of the CRH system. Moreover, we can subject these genetic mouse models to environmental challenges, which allow investigating mechanisms of gene × environment interaction.
A future challenge is to understand conditions and mechanisms of neuropeptide release and receptor activation in vivo. Finally, we aim at deciphering the signaling pathways downstream of CRHRs by disassembling the CRHR1 interactome and by investigating CRH/CRHR-dependent adaptations of the transcriptome and proteome. These efforts will advance our understanding of the organism's key stress system and will provide valuable insights to optimize CRHR-based treatment strategies.